

# 前言
探索反向折叠模型的前沿领域——这是一种创新而强大的工具,可改变从头蛋白质的设计格局。这些模型无缝地促进了治疗剂、生物传感器和工业酶的创造。加入我们的这篇博文,我们将深入探讨对反向折叠的全面理解,探索其应用,并揭示反向折叠模型的最佳利用方式,以实现无与伦比的蛋白质设计精度。
# 蛋白质折叠基础知识
让我们从蛋白质折叠开始。如果您不熟悉这个概念,蛋白质折叠是多肽或蛋白质优雅地转变为三维构象状态的过程,通常称为蛋白质的结构。先进的技术,如 AlphaFold2 和其他尖端的蛋白质结构预测模型 ,在这一领域发挥着关键作用。这些模型的工作原理是将给定蛋白质的氨基酸 (AA) 序列作为输入,并采用复杂的算法来预测其复杂的结构或折叠。这是一次引人入胜的旅程,探索控制这些基本生物构件形成的分子复杂性。
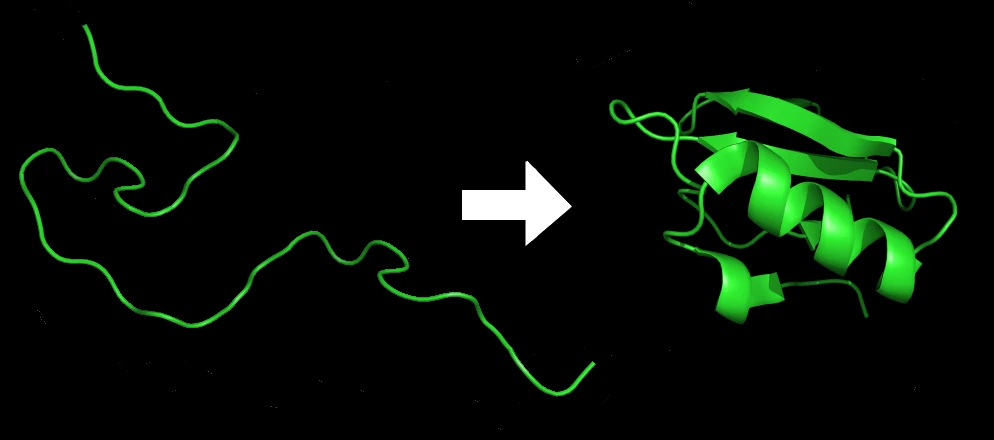
蛋白质折叠过程的图示
# 反向折叠基础知识
顾名思义,反向折叠与蛋白质折叠完全相反,它试图预测一个可以折叠成所需结构的序列。通常,这些模型是在大量掩蔽蛋白质结构数据集上训练的,然后模型必须预测原始序列。掩盖蛋白质结构是隐藏结构内发现的所有侧链的过程,以便仅保留蛋白质的骨架。这通常意味着唯一剩下的原子是对骨架至关重要的 α-碳、β-碳和氮。此外,一些模型(如 ProteinMPNN (opens new window))还会向这些原子的坐标制造一些影响或随机方差,以使模型更强大且更耐过度拟合 (opens new window)。这种掩蔽的蛋白质或蛋白质骨架也是模型在推理或预测阶段接收的输入。
虽然这似乎有悖常理,但对于蛋白质设计来说,这实际上是一个非常有用的过程,特别是对于酶和治疗剂。对于初学者来说,反向折叠模型往往非常快,可以预测数百个序列,这些序列可能在短短几分钟内折叠成所需的结构。此外,所得蛋白质之间的序列标识通常在 >0.4 到 <0.75 之间,这对于酶设计非常有价值,因为与可能仅依赖于几个点突变或插入缺失的传统方法相比,它允许您对序列空间的更广泛部分进行采样。
此外,像 ProteinMPNN 这样的模型是自回归的,并且与复合物配合得很好,允许您修复特定的链,这对于设计肽和小蛋白结合剂等疗法很有价值。例如,您可以使用 RFdiffusion (opens new window) 等工具来设计与靶蛋白或受体的特定区域相互作用的肽和微型结合剂。在此之后,可以使用反向折叠模型来创建序列,这些序列将产生所需的复合物,然后在后续步骤中对其进行筛选。
一些反向折叠模型(如 ProteinMPNN 和 ESM-IF1)的另一个好处是,它们带有置信度指标,可用于衡量预测的可靠性。
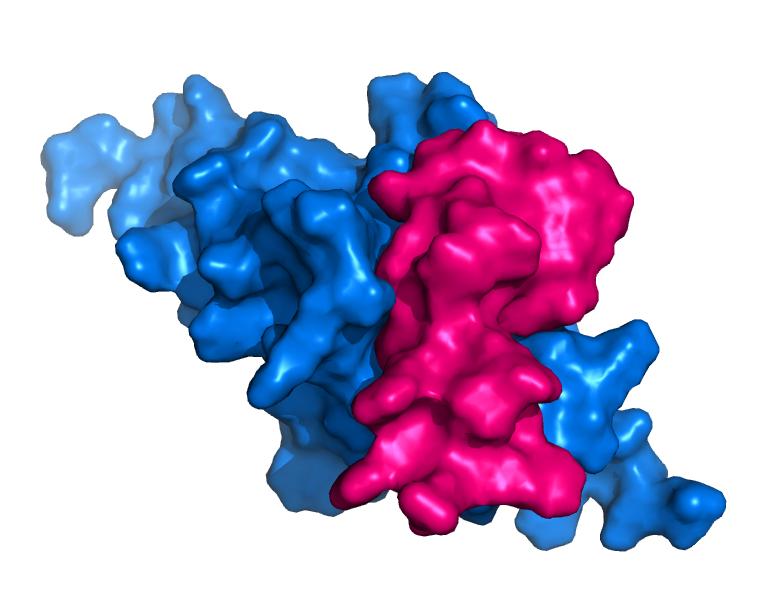
一种采用 AlphaFold2 设计的小型蛋白结合剂,用于抑制人PDCD1,作为治疗某些类型癌症的手段。